Vedi traduzione automatica
Questa è una traduzione automatica. Per vedere il testo originale in inglese cliccare qui
#Tendenze
{{{sourceTextContent.title}}}
Quando è entrata in vigore la norma 21 CFR Parte 11: cos’è e come garantirne la conformità
{{{sourceTextContent.subTitle}}}
La conformità al 21 CFR Parte 11 è obbligatoria per il settore farmaceutico e degli integratori alimentari regolamentato dalla FDA. Se non sapete di cosa si tratta e come ottenere tale conformità, troverete le risposte in questo articolo.
{{{sourceTextContent.description}}}
Nei settori manifatturieri regolamentati, la conservazione digitale dei dati non consiste più semplicemente nell’archiviazione di file. È diventata una fonte di prove tracciabili.
Per le aziende farmaceutiche, nutraceutiche, produttrici di dispositivi medici e di produzione conto terzi, i registri elettronici e le firme elettroniche sono essenziali per garantire la sicurezza della produzione, mantenere la garanzia di qualità e prevenire costose interruzioni causate da modifiche alle formulazioni o da adeguamenti dei parametri di processo. Con l’evoluzione delle normative statunitensi, la conformità al 21 CFR Parte 11 è diventata gradualmente un requisito obbligatorio piuttosto che un investimento facoltativo.
Per molti produttori, in particolare le piccole e medie imprese, l’implementazione della convalida dei sistemi e di soluzioni di gestione dei dati conformi può sembrare costosa e complessa. Di conseguenza, spesso vengono poste innanzitutto due domande fondamentali: quando è entrata in vigore la norma 21 CFR Parte 11 e in cosa consiste la conformità alla norma 21 CFR Parte 11?
Questa guida completa offre una spiegazione dettagliata del 21 CFR Parte 11. Esploreremo cos’è il 21 CFR Parte 11, chi è tenuto a conformarsi ad esso, i suoi requisiti chiave e come raggiungere la conformità al 21 CFR Parte 11 in modo pratico ed economicamente vantaggioso.
Punti chiave
Quando è entrata in vigore la norma 21 CFR Parte 11 e qual è il suo contesto storico.
Che cos’è la norma 21 CFR Parte 11 nell’industria farmaceutica.
Chi deve garantire la conformità alla norma 21 CFR Parte 11 nei vari settori industriali.
Requisiti fondamentali del 21 CFR Parte 11 relativi a registrazioni, firme, accesso e audit trail.
Come conformarsi al 21 CFR Parte 11 passo dopo passo.
In che modo le apparecchiature conformi alla Parte 11 supportano la produzione farmaceutica e nutraceutica.
1. Quando è entrata in vigore la norma 21 CFR Parte 11?
Secondo il sito web ufficiale della FDA, la norma 21 CFR Parte 11 è entrata in vigore il 20 agosto 1997.
Cosa ha portato all’emanazione di questa normativa? Le sue origini risiedono nella grande transizione dai registri cartacei ai sistemi informatizzati. In precedenza, i registri di produzione farmaceutica, i rapporti di laboratorio e i documenti relativi alla qualità venivano in gran parte redatti, firmati e archiviati su carta. Con l’espansione delle scale di produzione, i semplici registri cartacei non erano più in grado di soddisfare l’esigenza di una documentazione rapida e di un recupero immediato dei dati, portando le aziende ad adottare sistemi digitali.
Per garantire l’affidabilità di questi nuovi sistemi, la Parte 11 del Titolo 21 del Codice dei regolamenti federali (21 CFR Parte 11) sottopone i registri elettronici e le firme elettroniche a una rigorosa supervisione normativa. Quando le aziende utilizzano dati digitali a supporto di decisioni soggette alla regolamentazione della FDA, tali dati devono essere accurati e sicuri, oltre che tracciabili e recuperabili durante le ispezioni della FDA.
2. Che cos’è il 21 CFR Parte 11?
Definizione del 21 CFR Parte 11
In sostanza, che cos’è il 21 CFR Parte 11? Si tratta di un regolamento stabilito dalla FDA statunitense che stabilisce le modalità di gestione dei registri elettronici e delle firme.
Per comprendere le norme del 21 CFR Parte 11, si pensi alla tradizionale supervisione esercitata dalla FDA sugli schedari chiusi a chiave, al controllo di chi li gestiva e alla tracciabilità di chi ne apriva o ne modificava il contenuto. Man mano che questi armadietti sono stati sostituiti da sistemi digitali, la FDA ha introdotto le linee guida del 21 CFR Parte 11 per garantire che i sistemi siano adeguatamente controllati e che i dati rimangano al sicuro.
Quali tipi di registrazioni sono interessati?
Le registrazioni elettroniche ai sensi del 21 CFR Parte 11 possono includere:
Registrazioni elettroniche dei lotti
Registrazioni di produzione
Registrazioni di controllo qualità
Registri di funzionamento delle apparecchiature
Risultati dei test di laboratorio
Documentazione relativa alla pulizia e alla manutenzione
Documentazione relativa alla formazione
Documentazione relativa al controllo delle modifiche e alle deviazioni
Va notato che questa normativa non si applica a tutti i file digitali; si applica solo quando la documentazione elettronica viene utilizzata per soddisfare i requisiti di conservazione dei dati previsti dalla FDA.
Quali settori devono conformarsi?
I requisiti di conformità previsti dal 21 CFR Parte 11 possono applicarsi a un’ampia gamma di soggetti, tra cui i produttori di prodotti farmaceutici, integratori alimentari e dispositivi medici, nonché alle organizzazioni di produzione a contratto, ai laboratori di analisi e alle aziende che esportano negli Stati Uniti prodotti regolamentati dalla FDA.
Di conseguenza, domande quali «Il 21 CFR Parte 11 si applica ai dispositivi medici?» e «Cosa copre il 21 CFR Parte 11?» sono fondamentali; il principio fondamentale che regola l’applicabilità della norma è che i requisiti di conformità si applicano ogni volta che si utilizzano sistemi elettronici a supporto di registrazioni o approvazioni soggette ai requisiti di integrità dei dati della FDA.
3. Perché è stato introdotto il 21 CFR Parte 11?
Il 21 CFR Parte 11 è stato introdotto perché la produzione digitale ha generato sia nuovi vantaggi in termini di efficienza sia nuovi rischi di non conformità.
Il 21 CFR Parte 11 è nato in risposta ai guadagni in termini di efficienza e ai nuovi rischi di conformità associati alla produzione digitale. Man mano che il settore passava dai registri cartacei ai sistemi digitali, la FDA ha dovuto stabilire norme che garantissero l’affidabilità dei registri elettronici; questa norma si applica ai registri regolamentati dalla FDA che vengono creati, modificati, conservati, archiviati, recuperati o trasmessi elettronicamente.
L’obiettivo principale della norma è salvaguardare l’integrità dei dati ai sensi del 21 CFR Parte 11 e prevenire manomissioni o falsificazioni, richiedendo che i sistemi dispongano di audit trail, che fungono in modo molto simile a una “scatola nera” che registra la sequenza reale degli eventi. Durante le ispezioni della FDA, le autorità di regolamentazione esaminano i registri elettronici dei lotti, i requisiti relativi alle tracce di audit previsti dalla Parte 11, i controlli di accesso degli utenti e le firme elettroniche conformi al 21 CFR Parte 11; questi registri storici completi costituiscono la base fondamentale per la conformità normativa, la qualità dei prodotti e la fiducia dei clienti.
4. Chi deve garantire la conformità al 21 CFR Parte 11?
Non tutte le aziende necessitano dello stesso livello di controllo, ma molti produttori soggetti alla regolamentazione della FDA dovrebbero valutare chi deve conformarsi al 21 CFR Parte 11 in base ai propri sistemi e al proprio mercato.
Produttori di farmaci
Nel settore farmaceutico, i sistemi di registrazione elettronica che riguardano informazioni sui lotti, il controllo di qualità, i registri delle apparecchiature o le approvazioni di rilascio sono generalmente tenuti a conformarsi al 21 CFR Parte 11. Tuttavia, una volta che i registri elettronici sostituiscono formalmente quelli cartacei, tale obbligo di conformità diventa obbligatorio anziché facoltativo.
Produttori di integratori alimentari
Se un produttore di integratori alimentari gestisce elettronicamente le registrazioni relative alla produzione, alla qualità o ai lotti e il prodotto è destinato alla vendita negli Stati Uniti, è probabile che si applichi la conformità al CFR Parte 11.
Produttori di dispositivi medici
Per chi si chiede se il 21 CFR Parte 11 si applichi ai dispositivi medici, la risposta è sì, quando le registrazioni elettroniche soddisfano i requisiti del sistema di qualità della FDA.
Organizzazioni di produzione a contratto (CMO)
Le CMO necessitano spesso di una rigorosa conformità al 21 CFR Parte 11 della FDA poiché servono più clienti e sono soggette a frequenti audit da parte dei clienti.
Aziende che esportano negli Stati Uniti
Anche se il produttore ha sede al di fuori degli Stati Uniti, la conformità ai requisiti della Parte 11 è generalmente comunque richiesta, a condizione che i prodotti siano venduti negli Stati Uniti e che i registri elettronici riguardino attività regolamentate dalla FDA.
5. Quali sono i requisiti del 21 CFR Parte 11?
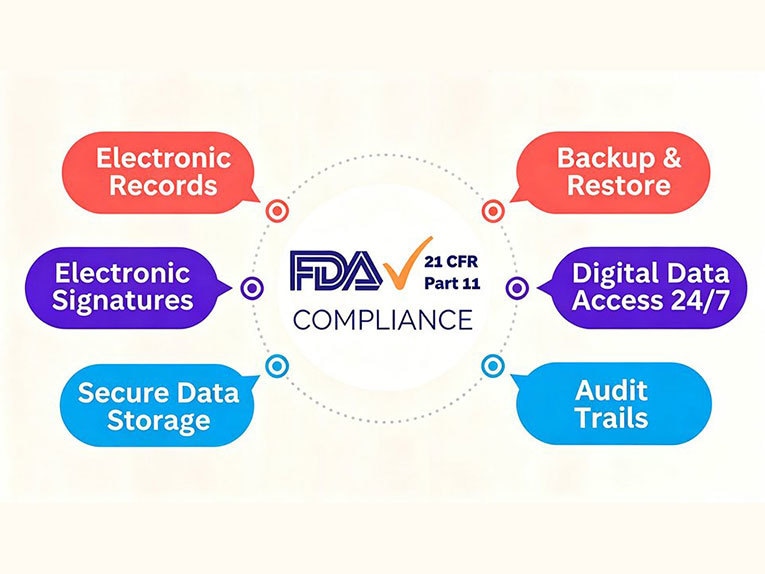
Ma, secondo il documento, quali sono esattamente i requisiti del 21 CFR Parte 11? La risposta può essere suddivisa in diverse misure di controllo pratiche. I requisiti di conformità previsti dal 21 CFR Parte 11 si concentrano su cinque aree chiave: registrazioni elettroniche, firme elettroniche, audit trail, controlli di accesso degli utenti e conservazione e recupero delle registrazioni.
Documentazione elettronica
I requisiti del 21 CFR Parte 11 relativi alla documentazione elettronica stabiliscono che i documenti elettronici debbano essere accurati, completi, sicuri, leggibili e recuperabili. Le aziende devono garantire un’adeguata conservazione dei documenti e un’archiviazione sicura dei dati.
Firme elettroniche
Le firme elettroniche previste dalla Parte 11 della FDA devono stabilire chiaramente l’identità e la responsabilità, riportando il nome del firmatario, il timestamp e il significato dell’azione (ad es. «approvazione per il rilascio») per garantire che abbiano la stessa validità legale delle firme autografe.
Registri di audit
Il funzionamento dei registri di audit nelle apparecchiature farmaceutiche è una domanda ricorrente. I registri di audit registrano automaticamente tutte le azioni critiche, tra cui creazione, modifica, cancellazione, approvazione e modifiche al sistema, per garantire la tracciabilità. Se i dati relativi al peso di un lotto vengono modificati da 500 kg a 510 kg, il registro di audit registra chi ha effettuato la modifica, l’ora della modifica e i valori prima e dopo la modifica, facilitando l’analisi delle cause alla radice.
Controlli di accesso degli utenti
I controlli di accesso garantiscono che solo il personale autorizzato possa eseguire azioni specifiche tramite protezione con password e gestione delle autorizzazioni, utilizzando l’autenticazione a più fattori e il principio del privilegio minimo.
Conservazione e recupero dei dati
I dati devono rimanere prontamente accessibili per tutto il periodo di conservazione previsto dalla FDA; la mancata recupero tempestivo dei dati durante un audit è considerata una grave violazione della conformità.
6. Come conformarsi al 21 CFR Parte 11
Il raggiungimento della conformità è una preoccupazione primaria per la maggior parte delle aziende farmaceutiche e di prodotti sanitari. In questa sezione approfondiremo l’argomento, trattando le fasi specifiche di implementazione, le potenziali problematiche e le relative soluzioni.
Fase 1 – Effettuare una valutazione di conformità al 21 CFR Parte 11
Si dovrebbe iniziare confrontando tutti i sistemi che creano, modificano, archiviano, recuperano o trasmettono registrazioni elettroniche regolamentate dalla FDA con le linee guida del 21 CFR Parte 11 per i prodotti farmaceutici. La valutazione dovrebbe includere la convalida, la traccia di audit, il controllo degli accessi degli utenti, le firme elettroniche, la conservazione dei dati e la sicurezza.
Seguire la checklist di conformità al 21 CFR Parte 11 può accelerare il processo.
Fase 2 – Convalidare i sistemi informatici
Un sistema conforme al 21 CFR Parte 11 deve essere sottoposto a convalida per dimostrare che funziona come previsto. In pratica, la convalida dovrebbe essere eseguita come una sequenza strutturata di azioni:
1. Definire e valutare l’ambito e il rischio del sistema
Iniziare documentando chiaramente ciò che il sistema deve fare e identificando dove si trova il rischio. Ad esempio, una macchina per il riempimento di capsule deve registrare il peso di riempimento, la velocità e il numero di lotto.
2. Testare e verificare le prestazioni del sistema
Successivamente, eseguire test strutturati utilizzando IQ, OQ e PQ. Questa valutazione serve a verificare se il sistema funziona come previsto.
3. Documentare, approvare e mantenere il controllo
Infine, tutte le attività di convalida devono essere documentate, riviste e approvate prima del rilascio del sistema.
Fase 3 – Implementare la funzionalità di audit trail
È necessario garantire l’integrità della funzione di audit trail, confermando che il sistema archivi e registri in modo sicuro i dati, quali i timestamp e le modifiche, e ne consenta un recupero accurato. Si prenda ad esempio la regolazione dei parametri della macchina riempitrice di capsule: una volta che un operatore modifica i parametri di riempimento, il sistema acquisisce automaticamente i valori precedenti e quelli nuovi, l’ID dell’operatore e il timestamp.
Fase 4 – Definizione dei controlli sulle firme elettroniche
Per soddisfare i requisiti della FDA in materia di firme elettroniche, ogni utente deve disporre di un’identità univoca. Le firme elettroniche devono essere collegate a record specifici e non devono essere copiate o riassegnate.
Fase 5 – Controllo dell’accesso degli utenti
Utilizzare un accesso basato sui ruoli. Ruoli diversi dovrebbero avere autorizzazioni diverse. Gli amministratori dispongono sempre delle autorizzazioni più elevate, mentre gli operatori hanno solo autorizzazioni parziali.
7. Ruida Packing Machinery: fornitura di macchine conformi alle norme FDA e GMP
A volte, anche se i produttori sono disposti a dotare le proprie macchine di un sistema di audit trail, non tutte le macchine lo supportano. Ruida Packing, in collaborazione con note aziende farmaceutiche, tra cui UCB e US Pharma, è indubbiamente impegnata nello sviluppo di apparecchiature conformi alla Parte 11. Le nostre macchine sono certificate CE, cGMP, ISO e dispongono delle relative certificazioni. In base alle esigenze dei clienti, Ruida è in grado di fornire anche soluzioni di monitoraggio conformi alla norma 21 CFR Parte 11 per le attrezzature di produzione e confezionamento.
Ruida supporta inoltre la produzione farmaceutica con servizi pratici e dettagliati:
Materiali certificati: acciaio inossidabile 304 per le parti non a contatto e acciaio inossidabile 316L per le parti a contatto.
Servizi di messa in servizio da remoto e in loco, con centri di assistenza post-vendita in Nord America e a Hong Kong.
Guida completa alle macchine: manuali, guide operative, documenti di manutenzione e video di assistenza post-vendita.
Documentazione IQ, OQ, PQ: supporto opzionale per i documenti di convalida (IQ, OQ, PQ) e la documentazione di conformità.
Per i produttori che intendono adeguarsi alla norma 21 CFR Parte 11, la scelta di apparecchiature dotate di opzioni già conformi può ridurre i costi di modifica futuri e facilitare la preparazione agli audit.
8. Conclusione
Guardando indietro a quando è entrata in vigore la norma 21 CFR Parte 11, possiamo vedere quanto profondamente abbia plasmato il settore dal 1997. Dal 20 agosto di quell’anno, questa normativa è stata lo standard di riferimento per la gestione dei dati elettronici da parte delle aziende soggette alla regolamentazione della FDA. Oggigiorno, la conformità al 21 CFR Parte 11 è al centro dell’attenzione e delle preoccupazioni di molti produttori del settore farmaceutico e degli integratori alimentari, ed è meglio prepararsi ad affrontarla. Non solo per quanto riguarda la convalida, ma anche per quanto riguarda le macchine.
9. Domande frequenti
D1: Il 21 CFR Parte 11 è obbligatorio?
Sì, certamente. Tuttavia, non si applica a tutti i sistemi di registrazione elettronica. È obbligatorio quando si utilizzano registrazioni elettroniche o firme elettroniche per soddisfare i requisiti della FDA.
D2: In cosa consiste la differenza tra le GMP e il 21 CFR Parte 11?
Beh, hanno obiettivi diversi. Le GMP si concentrano sulla qualità del prodotto, mentre la norma 21 CFR Parte 11 si concentra sulle registrazioni elettroniche e sulle firme elettroniche.
D3: Ogni macchina farmaceutica deve essere conforme alla Parte 11?
No. Una macchina deve essere valutata in base alla Parte 11 quando crea, archivia, modifica o trasmette registrazioni elettroniche soggette a regolamentazione. Le apparecchiature puramente meccaniche potrebbero non necessitare dello stesso livello di controllo.
D4: Che cos’è un audit trail?
Un audit trail serve a registrare tutte le azioni effettuate sul sistema, indicando l’azione esatta, l’ora e eventuali modifiche.
D5: Come faccio a sapere se un’apparecchiatura supporta i requisiti della Parte 11?
È possibile verificare se supporta le tracce di audit, il controllo degli accessi degli utenti, le firme elettroniche, l’archiviazione sicura dei dati, l’esportazione dei record e i documenti di convalida. Ma, in parole povere, basta chiedere ai fornitori delle apparecchiature prima dell’acquisto.
[1] U.S. Food and Drug Administration: Parte 11, Documentazione elettronica; Firme elettroniche — Ambito di applicazione e applicazione
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/part-11-electronic-records-electronic-signatures-scope-and-application
[2] Codice elettronico dei regolamenti federali: 21 CFR Parte 11 — Documentazione elettronica; firme elettroniche
[3] Registro federale: Documentazione elettronica; firma elettronica — Regola definitiva, pubblicata il 20 marzo 1997